Exactech Recall & Replacement Failure
Exactech has expanded the recall to include all knee and ankle arthroplasty polyethylene inserts packaged in non-conforming bags regardless of label or shelf life. During the period between August 2021 and January 2022, thousands of non-conforming knee and ankle devices have been shipped and implanted by surgeons.
There have been approximately 147,700 inserts implanted in the U.S. since 2004 that were produced with non-conforming packaging.
Three generations of Exactech knee systems have had polyethylene inserts packaged in non-conforming bags at various points during their respective market presence. The original Optetrak Knee system, from 1992, has shown higher overall revision rates.
Exactech ankle arthroplasty systems, or the Vantage® Total Ankle system, has been recalled for the same reasons as the knees. For patients who exhibit premature polyethylene wear, surgeons can consider revision surgery.
FDA has classified this field action as a class II recall, meaning that exposure to the product may cause medically reversable health consequences.
Many knee implant operations are successful and the devices function well for decades. But that is not always the case. Knee implant failure can be the result of a defectively designed component.
In the case of the Exactech Optetrak device, many reports indicate a loosening of the tibial tray, one of the device’s components linked to potential device failure.
The Optetrak component was deemed prone to excessive wear by the FDA and subject to a Class 2 device recall. Some patients who have received the Exactech implant have needed revision surgery to remove and replace the failed knee implant. Defective implants may not adhere fully and correctly to the bone, and lead to loosening, instability, pain, and infections.
Medical Device Recall & Injury
A study published in the medical journal Orthopaedics & Traumatology: Surgery & Research analyzed patients who were implanted with an Exactech total knee replacement system.
After about two years, the researchers found that tibial loosening was a common occurrence in the cement-tibial-implant component of the device, resulting in fragmentation. The implant failure was reportedly caused by tibial insert wear and tear of the implant.
Around 15 percent of patients said they were disappointed or dissatisfied with the implant. Over 20 percent of patients complained of Exactech knee pain that requires regular painkillers.
Around 22 percent were beginning to suffer from tibial implant loosening at only 25 months. Over a dozen of the reviewed patients needed revision surgery for tibial loosening, instability or pain. Revision surgery has its own risks of complications and infection.
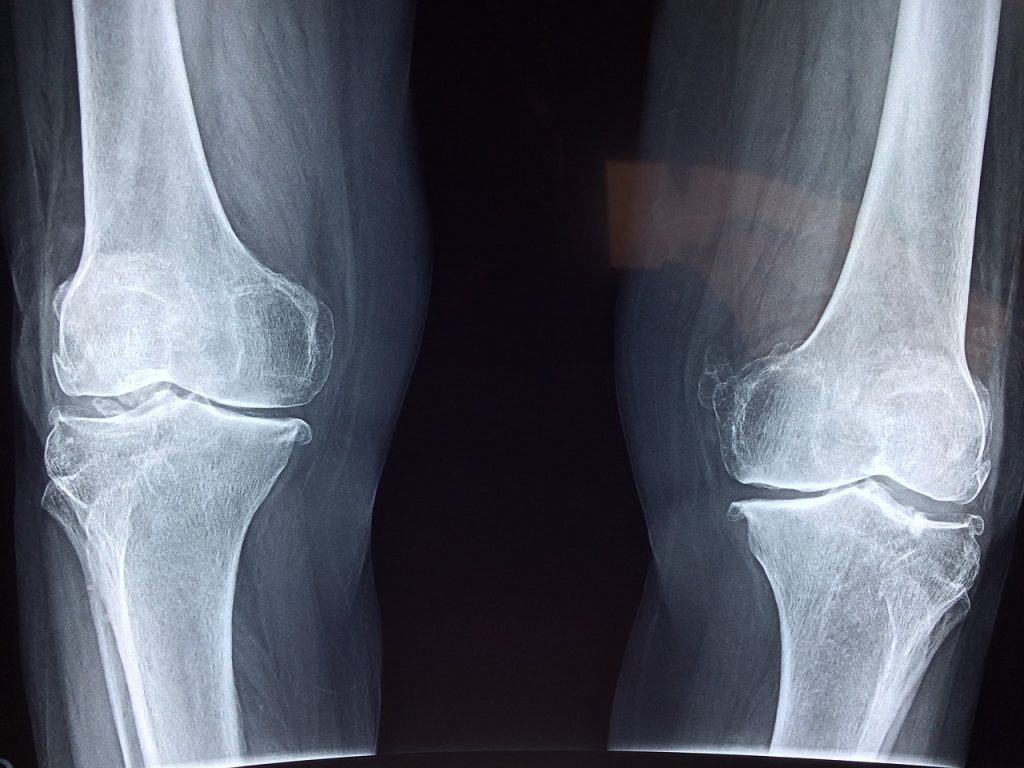
Exactech Knee Implant Recalls
The most recent extended Exactech recall involves knee and ankle implants, including the following models:
- Exactech Optetrak
- Exactech Truliant
- Exactech Vantage
A February 2022 Exactech implant recall was extended from an earlier 2021 recall due to improper packaging that allegedly impacted the safety and performance of the implants.
A study published in June, 2016, in the Journal of Arthroplasty concluded that some Knee implants may be associated with a high rate of baseplate loosening and failures that increase the risk of bone loss.
A related study published in the Journal of Knee Surgery said after reviewing patient and doctor reports, hospital databases, as well as independent studies, researchers encountered a high rate of debonding of the tibial implant–cement interface.
Patients with Exactech implants presented with pain on weight bearing and a decreased range of motion within 2 years after surgery. Numerous tibial failures were reported though researchers believed complications were underreported due to failure of radiographs to assess loosening.
Exactech Recall Lawsuits
When Exactech marketed their Optetrak product, the company promised patients a quick recovery time and a device manufactured to sustain harsh movements like exercise. Exactech has said the device was supposed to last longer and perform better than other knee implants on the market.
But many of the devices sold and implanted have been defective, and Exactech products have been causing further harm and injuries as a result. Patients who received Exactech knee implants were not expecting to undergo revision surgery for more than 15 years, making their early device failures a source of great distress and an important medical and legal matter. Complications associated with Exactech knee replacements include:
- Failure to bond
- Components wearing
- Fracture
- Implant detached from bone
- Persistent pain
- Bone Loss
- Infection
- Decreased range of motion
- Tissue Damage
- Nerve Damage
- Metallosis—metal debris shed into bloodstream
- Revision Surgery

Why are these cases important?
Defective medical device litigation helps to improve patient safety and holds medical device companies accountable when defective products injure plaintiffs and clients.
Companies may voluntarily recall their products, but the fact remains that much damage has already been done by the time some defective implants are recalled. Companies have a responsibility to properly test their devices before they are released to the public.